|
6月10日,国家药品监督管理局药品审评中心(CDE)发布了《先进治疗药品的范围、归类和释义(征求意见稿)》,首次从官方层面对细胞治疗药品(CTMPs)、基因治疗药品(GTMPs)等先进治疗药品(ATMP)进行了明确的定义、归类及分类原则。这标志着我国细胞与基因治疗领域正式迈入精准监管时代,也预示着一个行业大发展的窗口正在打开。
一、新政出台的背景与目的
近年来,干细胞疗法、基因治疗、CAR-T细胞疗法等先进治疗技术迅猛发展,相关创新产品不断涌现,并在癌症、罕见遗传疾病等疑难重症治疗领域实现了突破性进展,多个先进治疗药品陆续进入临床试验或获得上市批准。当前,我国细胞治疗产业已进入与国际先进水平“并跑”的新阶段。

然而,我国尚缺乏统一明确的监管标准,导致各类产品的审批和注册过程存在较大的不确定性。研发企业、监管机构乃至患者,在产品的安全性、有效性以及质量控制方面都存在诸多困惑。为此,国家药监局药品审评中心组织起草了本征求意见稿,其主要目的在于:规范先进治疗药品的定义与范围,消除监管模糊地带;实现分级分类监管,更加精准高效;推动我国监管标准与国际接轨,促进国际合作;加速创新药物上市,满足患者迫切需求。
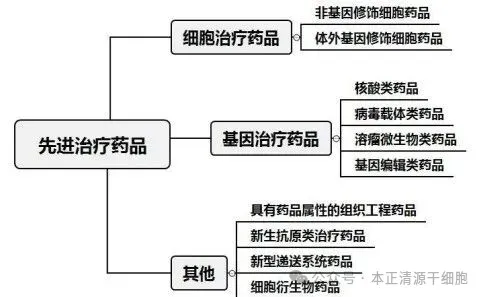
二、明确分类:三大类,写进官方文件
新政将先进治疗药品分为三大类:
细胞治疗药品(CTMPs): 包括干细胞、NK、TIL未经或经体外基因修饰的CAR-T、CAR-NK等,以细胞为基础,通过再生、替代或免疫调节发挥疗效。
基因治疗药品(GTMPs): 利用DNA、RNA或病毒等载体,在体内实现基因序列或基因表达的调节,如rAAV载体药物、mRNA药物、CRISPR基因编辑药物等。
其他先进治疗药品: 涵盖组织工程药物、人造组织、外泌体药物等创新形态药物,为未来新兴技术预留空间。
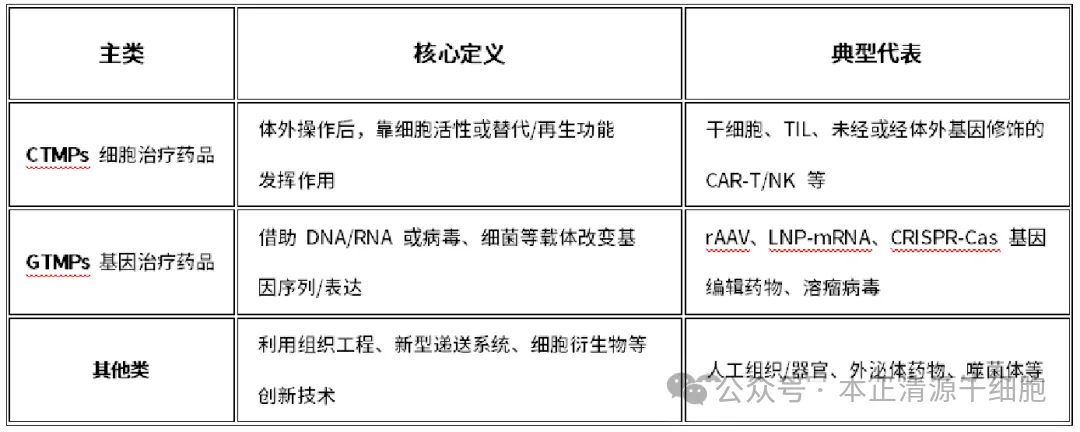
值得注意的是,政策特别强调:细胞疗法被归类为“体外基因修饰的细胞治疗药品”,而非国际上部分地区将其归为基因治疗药品的做法,这充分体现了政策制定的科学性与灵活性。
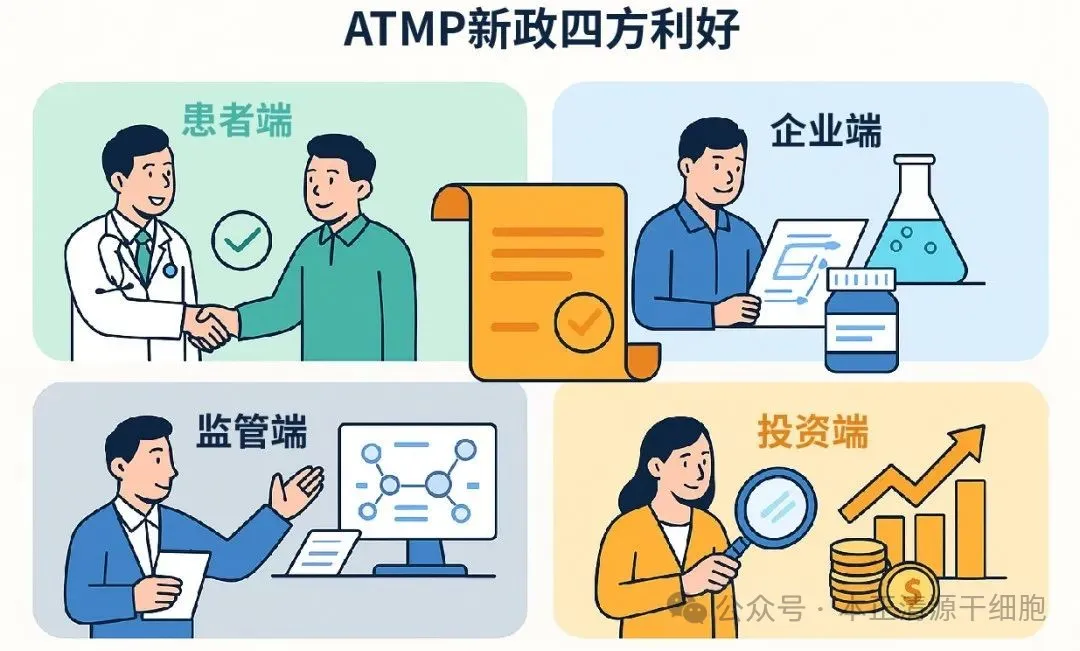
三、新政将带来的多方利好
新政策的落地,对于行业各方都将带来积极影响:
患者端: 产品审批加速,药品质量与安全性更有保障,患者获得治疗的途径将更加通畅。
企业端: 明确注册类别与申报路径,降低研发成本与风险,促进企业创新活力。
监管端: 明确分级分类监管标准,有利于高效审批并减少监管负担,进一步提高监管科学性。
投资端: 明确的政策导向有助于资本市场理性投资决策,促进资金进入真正有前景的研发项目。
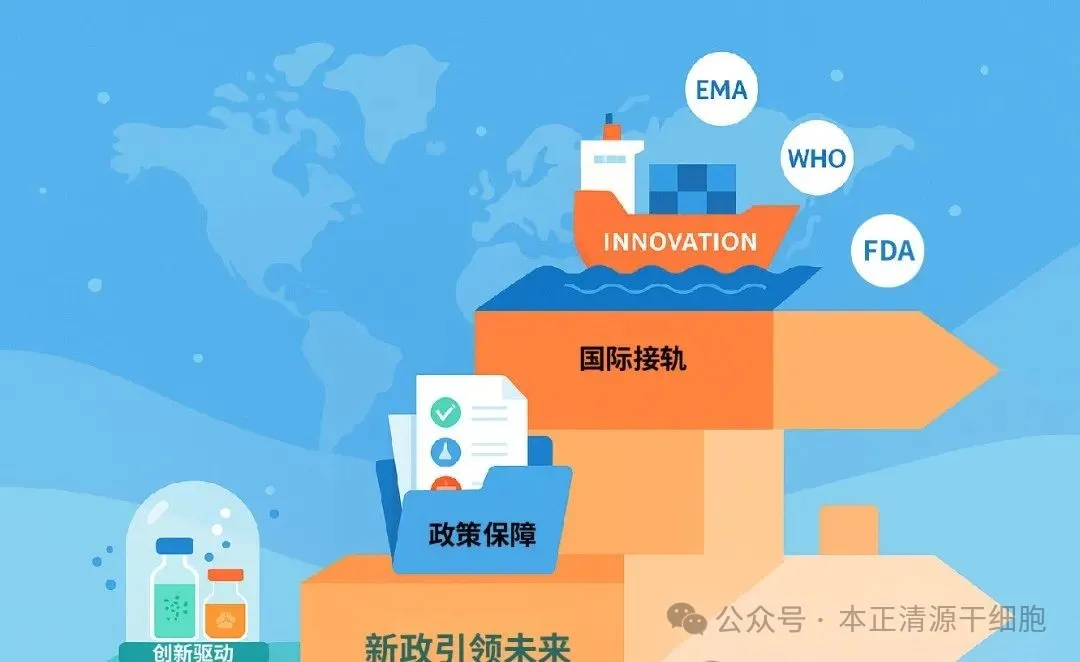
四、开启产业发展的新纪元
从更长远来看,这一新政的出台将从以下几个维度推动产业高质量发展:
实现国际化接轨: 新政与EMA(欧洲药品管理局)的ATMP分类、FDA的CGT监管体系高度对标,有利于促进国内产品参与国际多中心临床试验,加速创新药物全球上市。
推动配套政策落地: 新政为未来出台更具体的技术指导原则、伦理与生物安全细则、医保、商保、患者援助支付与准入政策提供了明确的基础,全面提高产品可及性。
加速技术创新落地: “其他类”明确了新型递送技术、外泌体、组织工程药物等未来方向,鼓励企业大胆探索。
五、结语:新政助力,细胞与基因疗法驶入发展快车道
本次征求意见稿的出台,标志着我国细胞与基因疗法监管正式进入精准化时代。这不仅是行业的一个里程碑,更将成为患者福音、企业创新的助推器、资本市场投资的风向标。
从此,干细胞、基因治疗、外泌体等前沿疗法将告别“摸着石头过河”的时代,迎来更快的审批通道、更清晰的发展路径和更安全的医疗选择。期待更多业界人士积极参与政策完善,推动中国细胞与基因治疗产业迈向更广阔的未来。
|






